Effect of Microcrystalline Cellulose Particle Size on Flowability and Compressibility in
Direct Compression Method
Dr. Viraj Vithal Kulthea*, Gunjan Manish Kshirsagara
, Amit Raj Sinhab
aSIGACHI Industries Limited, Dahej SEZ – I, Bharuch-392130, Gujarat, India.
bSIGACHI Industries Limited, 4th Floor, Kalayan’s Tulsiram Chambers, Madinaguda, Hyderabad-500049, Telangana, India
SIGACHI Industries Limited. Unit – 2: Plot No. Z-16, Dahej SEZ, Part – 1 Dahej, District – Bharuch, Gujarat-392130, India
SIGACHI Industries Limited. Unit – 2: Plot No. Z-16, Dahej SEZ, Part – 1 Dahej, District – Bharuch, Gujarat-392130, India
SIGACHI Industries Limited. 4th Floor, Kalayan’s Tulsiram Chambers, Madinaguda, Hyderabad, Telangana-500049, India
ABSTRACT
Microcrystalline cellulose is used as a tablet diluent and binder. It also imparts lubrication and self-disintegration properties to the
formulation that makes it a multifunctional excipient in tableting by both wet-granulation and direct-compression methods. It is
commercially available in different particle sizes, densities and moisture levels that define its different applications. This study aims
at evaluating influence of particle size of microcrystalline cellulose on flow and compressibility properties of diclofenac in
manufacturing oral tablets by direction compression method. This study demonstrated that out of 4 different grades of
microcrystalline cellulose viz.25, 50, 100 and 200 μm, the grade with 200 μm particle size distribution (HiCelTM LP200) showed
superior powder micromeritics to other grades. Whereas the grade with 50 μm particle size distribution (HiCelTM 50M) showed
superior tablet characteristics and performance to other grades. This suggested that the particle size of microcrystalline cellulose
influenced blend mixing uniformity, flow and density, filling of void spaces in die cavity and compressibility into tablets. This was
evident with excellent angle of repose, Carr’s index and Hausner’s ratio of diclofenac blend containing HiCelTM LP200. The
diclofenac blend containing HiCelTM 50M produced tablets with better strength, least weight variations and quick disintegration as
compared to the other grades. On the contrary, diclofenac blend containing fine grade of microcrystalline cellulose (HiCelTM 25M)
showed poorer micromeritics and produced tablets with unacceptable strength and weight variations. Thus, particle size of
microcrystalline cellulose is decisive in ensuring good processability, product quality and performance.
Keywords: microcrystalline cellulose, particle size distribution, direct compression, tablet properties, powder micromeritics.
1. Introduction
Tablets are solid dosage forms in which one or more APIs are blended with excipients and compressed into the final formulation.
Conventionally, tablets remain the preferred dosage form due to the advantages, like ease of manufacturing, dosage accuracy, longer
shelf life, high patient acceptability and compliance (1). The simplest manufacturing technique, direct compression is acceptable
only when the APIs and excipients possess acceptable flow and compression properties without additional processing steps. Belief
in advanced commercialisation approaches also, like continuous manufacturing in pharma industry is growing stronger because it
addresses the practical challenges in conventional batch manufacturing, such as increased product handling, more manufacturing
steps, constraints due to higher plant footprint, time-consuming changeover, and quality control (2). It is a well-known fact that bulk
powders inherently exhibit unpredictable flow in feeders, dosing machines, packing machines under the influence of gravity. Such
manufacturing processes are significantly influenced by the quality of input materials along with other aspects. Majority of the
formulations contain excipient concentration higher than API. Thus, it mandates appropriate selection of starting materials with
excellent micromeritics.
Nofrerias et al proposed characterization of pharmaceutical powders by means of the SeDeM diagram expert system. Their study
comprised of the characterization of Microcrystalline Celluloses (MCC) of different grades (101, 102, 301, 302 and 200) made by
four different manufacturers. The SeDeM characterization led to describing and to analysing the differences between each MCC
studied based on dimensions, compressibility, flowability, lubricity/ stability and lubricity/ dosage) and the Index of Good
Compression (IGC). They concluded that although the MCC manufacturers usually describe similarly in their products, the results
indicated that there were large differences between them based on above parameters (3). Ouazzou et al have demonstrated the
influence of the particle size distribution of maltodextrin powders, the compression pressure and the dwell time on the tensile strength, porosity, and pore size distribution of the final tablet produced by wet granulation technique (4). Wünsch et al had
systematically characterized the influence of initial particle size on powder compressibility and compactibility by consideration of
in-die compressibility, specific energies, quick elastic recovery, tablet porosity and, tensile strength for the binder microcrystalline
cellulose and three APIs. They concluded that the compactibility was increased with decreasing particle size because of the
increasing number of bonds in a cross-sectional area of the tablet, as found by the application of the model of Rumpf (5).
Microcrystalline celluloses, because of their multifunctional properties like filler, binder, disintegrant and self- lubrication are
commonly used in direct compression tablet formulations. Physical properties of such excipients, like density, crystal habit, moisture
content, particle size, shape, porosity, static charges and morphology have decisive impact over processability, physicochemical
quality attributes, stability, performance and overall therapeutic success of the formulation (6).
The viscoelastic behavior of microcrystalline cellulose also demonstrates greater elastic effects at higher tableting speeds where
there is insufficient compaction time for plastic deformation (7). Microcrystalline cellulose has a very high intraparticle porosity
with approximately 90–95% of the surface area being internal. Therefore, surface area is not directly influenced by the nominal
particle size. The plasticity of microcrystalline cellulose is the main reason of its exceptional binding properties. However, compared
to brittle excipients, microcrystalline cellulose is more lubricant sensitive. Lubricated microcrystalline cellulose particles will
deform under pressure and will not fracture to create new and clean (lubricant-free) surfaces (8). The presence of high levels of
hydrophobic lubricants, such as, magnesium stearate, the use of long blend times and high blend speeds would then result in softer
tablets (9). The addition of brittle excipients or colloidal silicon dioxide into the blends containing microcrystalline cellulose can
successfully prevent lubricants from occupying the microcrystalline cellulose surfaces, and in turn minimize the negative influence
of lubricants on tablet strength. Microcrystalline cellulose has a very high intraparticle porosity with approximately 90–95% of the
surface area being internal, this high porosity promotes swelling and disintegration of microcrystalline cellulose tablets by
penetration of water into the tablet matrix.
Hence, the influence of particle size distribution as key material attribute of microcrystalline cellulose on processability, quality
attributes of powder blend and tablets, and their performance was studied in the present study. Different varieties of spray dried
microcrystalline cellulose having particle size distribution ranging from 25- 200 μm with specific densities were used to formulate
the pharmaceutical tablets by direct compression technique. The physical blends were evaluated for flow, compressibility and other
micromeritics. Out of several formulations, the blend containing coarser grades of microcrystalline cellulose showed better powder
micromeritics. Further, the pharmaceutical tablets were evaluated mainly for strength, disintegration and dissolution. Out of several
formulations, the tablets containing finer grades of microcrystalline cellulose showed better performance in terms of quicker
disintegration and complete dissolution. Thus, it is possible to conclude that the particle size distribution of the major components
in tablet formulations has decisive impact over, powder physical properties, processability tablet properties and its performance
also.
2. Experimental
2.1 Materials
HiCelTM 25M, HiCelTM 50M, HiCelTM 90M, HiCelTM LP200 were manufactured at SIGACHI® Industries Limited, Dahej, Gujarat,
India. Diclofenac sodium was purchased from Pharma Lab Limited, India, colloidal silicon dioxide was purchased from Evonik
Industries Germany, Croscarmellose sodium was purchased from Sigachi Industries Limited, Dahej, Gujarat, India, Purified talc
powder was purchased from Neelkanth Fine Chem LLP, Jodhpur, Rajasthan, Magnesium Stearate was purchased from Sunshine
Organic PVT. LTD. Mumbai, India.
2.2 Methods
2.2.1 Manufacturing of diclofenac sodium tablets by direct compression method
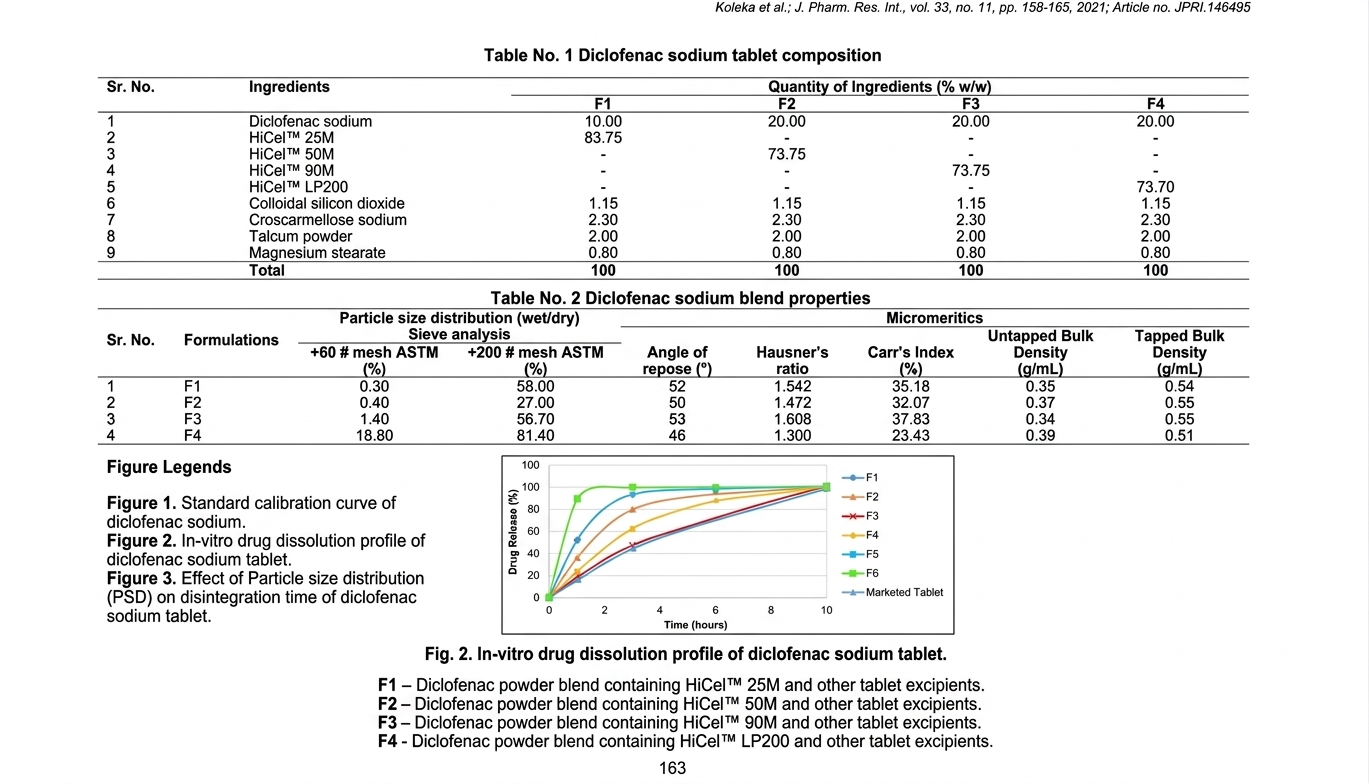
The diclofenac sodium tablet formulations as detailed in the Table No.1, strategically incorporated four distinct grades of HiCelTM
(Microcrystalline Cellulose) alongside other critical excipients to achieve desired tableting characteristics and drug delivery profiles.
Each HiCelTM grade is selected based on its unique micromeritic properties, which dictate its functional role within the tablet matrix.
Supplementary excipients, such as binders, disintegrants, lubricants, and glidants, were selected to collectively optimize powder
flow, compaction behavior, tablet integrity, and performance of the final dosage form. Accurately weighed quantities of diclofenac
sodium, croscarmellose sodium, microcrystalline cellulose, and colloidal silicon dioxide were co-sifted through a 30# ASTM
stainless steel sieve. They were transferred into powder blender of appropriate capacity (Make: Reva Pharma Machinery, India,
Model: TRMIX-20). These materials were mixed for 15 min at 25 rpm to ensure blend homogeneity. To this premix, an accurately
weighed quantity of talcum powder (previously shifted through a 40# mesh ASTM sieve) was added and mixed for 15 min at 25
rpm. The entire mixture was then subjected to final sifting through a 30# mesh ASTM sieve. Finally, magnesium stearate, previously
sieved through a 60# mesh ASTM sieve, was added into the blended material and mixed for 2 min to facilitate lubrication. Then,
this final lubricated blend was compressed into tablets by using a 12-station tablet compression machine (Make: Eliza Press, India,
Model: EP-200) fitted with 9.50 mm diameter standard rounded shaped “D” tooling set.
2.3 Evaluation of Pre-Compression Parameter
The final lubricated blend was characterized by angle of repose, untapped bulk density, tapped bulk density, Hausner’s ratio and
compressibility index parameters.
2.3.1 Untapped bulk density
Untapped bulk density was analyzed by using volumeter by the method mentioned in United States Pharmacopoeia: General Chapter
on bulk density <616> (10).
2.3.2 Tapped bulk density
Tapped bulk density was determined by the method mentioned in United States Pharmacopoeia: General Chapter on tapped density
<616> (10), using density apparatus (Make: Electrolab, India Model: USP 1&2).
2.3.3 Hausner’s ratio
It is an indirect index of measuring powder flow by the method mentioned in United States Pharmacopoeia: General Chapter on
powder flow <1174> (11), using a tap density apparatus (Make: Electrolab, India Model: USP 1&2).
2.3.4 Angle of Repose
It is an indirect index of measuring powder flow by the method mentioned in United States Pharmacopoeia: General Chapter on
powder flow <1174> (11)
2.3.5 Compressibility index (Carr’s index)
It is an indirect index of measuring powder flow by the method mentioned in United States Pharmacopoeia: General Chapter on
powder flow <1174> (11). Lower Carr’s index (between 15 and 20%) indicated fair compressibility property of the MCC.
2.3.6 Sieve analysis
Sieve analysis was performed by arranging different types of mesh sieves by the method mentioned in United States Pharmacopoeia:
General Chapter on powder flow <786> (12).
2.4 Evaluation of tablet properties
2.4.1 Weight variation
Tablet weight variation was evaluated as per United States Pharmacopoeia: General Chapter on Uniformity of dosage units <905>
(13).
2.4.2 Tablet Compaction
Compacts of 250 mg tablet in tablet compression machine 12 stations, with “D” tooling set (Make: Eliza Press, India, Model: EP-
200). Machine operating pressure ranges 10 to 60 kN.
2.4.3 Tablet Hardness
Tablet hardness was determined using a calibrated hardness tester (Make: Labindia tablet hardness tester, India, Model No.-TH1050
M) as per the United States Pharmacopoeia: General Chapter <1217> (14).
2.4.4 Tablet Thickness
Thickness was measured by using a digital tablet thickness tester machine (Make: Labindia tablet hardness tester, India, Model No.-
TH1050 M).
2.4.5 Friability
Tablet friability test was performed by using the friabilator (Make: Labindia India, Model: FT1020). The percentage friability was
determined by method mentioned in United States Pharmacopoeia: general chapter on tablet friability <1216> (15).
2.4.6 Disintegration
Tablet Disintegration Tester machine (Make: Labindia, India, Model: DT1000) was used for evaluating disintegration time as per
United States Pharmacopoeia: general chapter on disintegration time <701>, disintegration time of uncoated tablets should be NMT
15 min (16).
2.4.7 In-vitro dissolution test
The dissolution profile of diclofenac sodium immediate release tablet was performed using dissolution test apparatus (Make:
Labindia India, Model: DS8000) by using the USP 44 compendial method, apparatus II (paddle), speed 50 rpm for 1 hr in 900 mL
of phosphate buffer pH 7.5 at 37 ± 0.5°C medium temperature. Six tablets were placed in each dissolution vessel. Sample aliquots
of 5 mL were withdrawn from each dissolution vessel at 5, 15, 30, 45 and 60 min. After withdrawal, samples were filtered through
whatman filter paper (No. 45). 1 mL of filtrate from the beaker was taken and transferred into 10 mL of volumetric flask and diluted
up to 10 mL with dissolution medium. The same procedure was repeated for all remaining 5 tablets. Standard and sample
absorbances were recorded using a UV spectrophotometer (Make: Shimadzu Japan, Model: UV-1900) at λ= 276 nm wavelength.
The diclofenac sodium released from tablet formulations was calculated using the below mentioned formula (17).
Amount of drug released (mg) = Concentration of released drug × Dilution factor ×Volume of dissolution medium / 1000
Drug dissolved (%) = Amount of drug released (mg) / label claim (mg) × 100
2.4.7.1 Calibration curve of diclofenac sodium tablet
Phosphate buffer pH 7.5 filtered through whatman filter paper (No. 45) was used as diluent for preparing the standard solution of
the drug. About 10 mg of diclofenac sodium was accurately weighed and transferred into 100 mL volumetric flask, the solution was
sonicated and the resulting solution was diluted with the diluent to obtain a primary stock solution to obtain 100 μg/mL diclofenac
sodium. From this solution, suitable standard solutions of different concentrations were prepared. Linearity of the absorbance was
determined by measuring standard dilution of diclofenac sodium in the range of 5 – 20 μg/mL. The absorbance of diclofenac sodium
was monitored at 276 nm and the corresponding absorbance were obtained. From those absorbances, a graph of concentration and
absorbance was plotted. The regression equation (Figure 1) obtained was used to estimate the amount of drugs present in tablet
dosage formulations.
3. Results and discussion
3.1 Powder micromeritics
Diclofenac sodium formulations were prepared by using four grades of HiCelTM samples. These powder blends were evaluated by
physical tests as in Table No. 2. Formulation F1, contained HiCelTM 25M, exhibited poor flowability, evidenced by an angle of
repose of 52° and a bulk density of 0.35 g/ mL. This indicated a highly cohesive powder, posing potential challenges for uniform
die filling during tablet compression (18). In contrast, formulations F2 and F3, containing HiCelTM 50M and HiCelTM 90M
respectively showed improved flow characteristics. F2 presented an angle of repose of 50° and a bulk density of 0.37 g/ mL, while
F3 displayed an angle of repose of 53° and a bulk density of 0.34 g/ mL. F3 shows higher angle of repose, both grades were generally
recognized for their favorable flow properties, suggesting a balance of particle size and interparticulate forces conducive to more
consistent powder handling. Formulation F4, which comprised the HiCelTM LP200 grade, exhibited superior flowability,
characterized by a lower angle of repose of 46°. This indicated a highly free-flowing powder blend, attributable to the coarser
particle size distribution typical of HiCelTM LP200.
3.2 Tableting properties
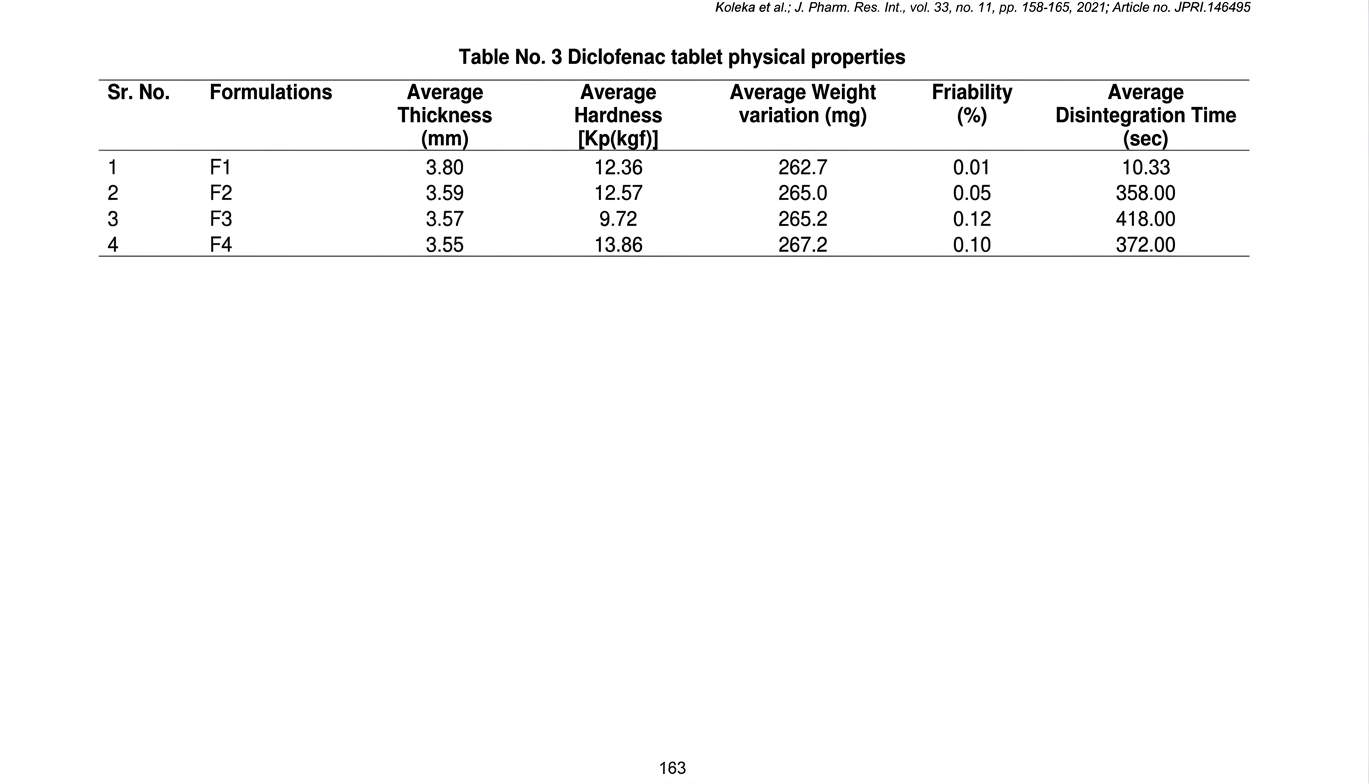
Physical properties of diclofenac sodium tablet containing varying grades of HiCelTMand obtained by direct compression technique
is represented in Table No. 3. Formulation F1, contained HiCelTM 25M, exhibited good compressibility. This was primarily
attributed to the fine particle size of HiCelTM 25M, which facilitated completely filling interparticulate void spaces, intimate particle
contacts and enhanced interparticulate bonding upon compression. Furthermore, the inherent high surface area of these fine particles
resulted into quick disintegration time, it enabled efficient water wicking properties and subsequent rapid tablet breakdown (19).
Conversely, formulations F2 and F4, containing HiCelTM50M and HiCelTM LP200, showed comparable hardness and disintegration
time indicated that both grades provided a favorable balance between tablet mechanical strength and water adsorption. The coarser
nature of HiCelTM LP200 (in F4) typically provided superior flow, which combined with its established binding properties,
contributed to robust tablets (20). Formulation F3, featuring HiCelTM 90M, showed a relatively lower tablet hardness and a
prolonged disintegration time of 418 sec. Also exhibiting higher friability than all other formulations. The overall performance of
F3 in terms of hardness and disintegration was less favorable compared to F2 and F4. This suggests that while HiCelTM 90M offered
adequate binding and higher friability, its particle size and characteristics may have resulted in a less efficient compaction profile
compared to the other grades, thus influencing the critical quality attributes of the diclofenac sodium tablets.
3.3 In-vitro Dissolution study of diclofenac sodium immediate release tablet
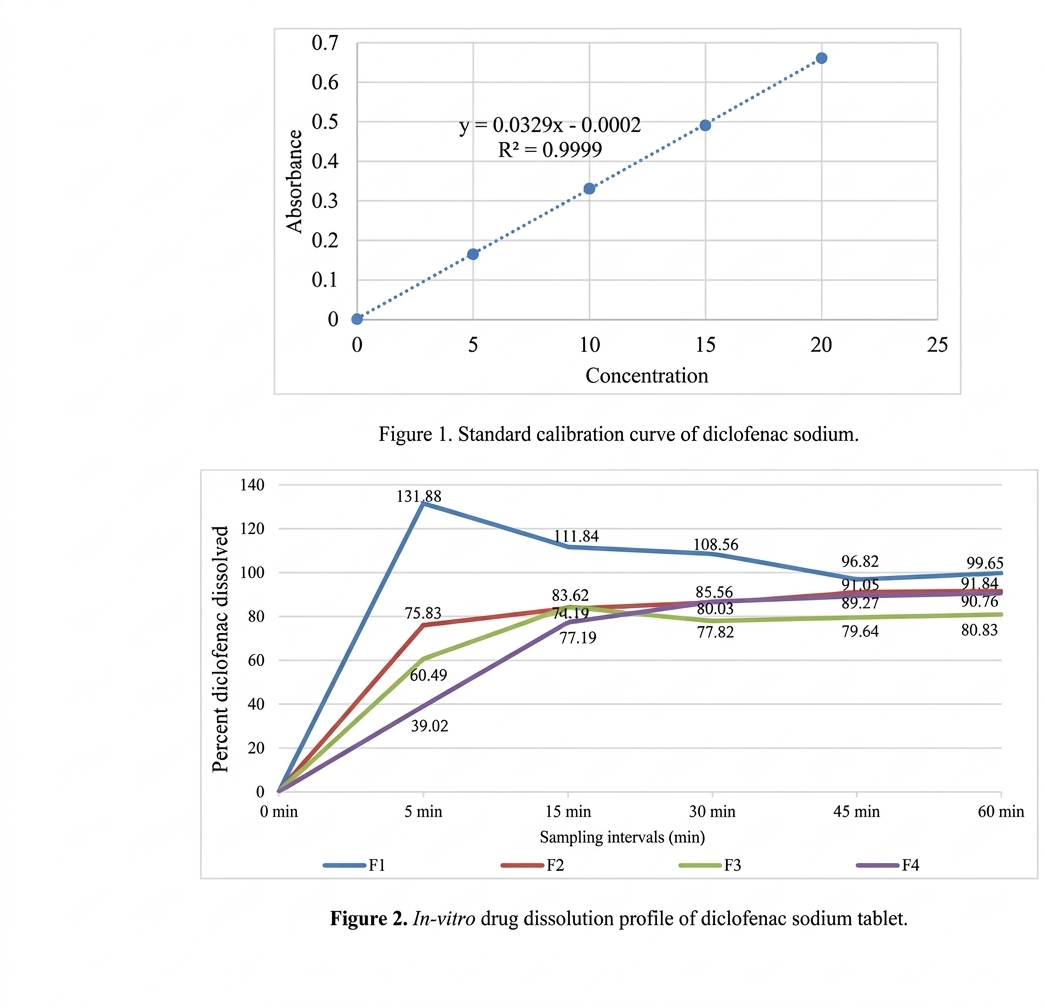
At least 80% of the labeled amount of diclofenac sodium should be dissolved within 60 min (21). A comprehensive dissolution
study was conducted on diclofenac sodium tablets, incorporating four distinct grades of HiCelTM Microcrystalline Cellulose (MCC)
powder. As showed in Figure 2, F1 formulation which contained HiCelTM 25M that is finer grade of microcrystalline cellulose,
exhibited an exceptionally rapid and complete drug dissolution. Drug release at the initial 5 min interval, followed by 111.84% at
15 min, 108.56%. This accelerated dissolution profile was attributed to the high surface area of fine particle of the HiCelTM 25M.
This might have contributed to an increased surface area available for dissolution (22, 23). However improper filling of die cavity
due to poor flowability of fine particles lead to fluctuation or unevenness in uniform distribution of powder blend resulting in uneven
drug assay, disintegration and dissolution. Conversely, the remaining three formulations (F2, F3 and F4) demonstrated dissolution
results well within acceptable compendial specifications. Notably, the F4 formulation, containing HiCelTM LP200, exhibited a
superior drug dissolution profile as 90.78% drug dissolution were the other, F2 showed 91.84%, F3 showed 80.83% drug dissolution
that closely aligned with the requirements stipulated in the United States Pharmacopeia (USP) monograph for diclofenac sodium
tablets. All tested formulations successfully achieved more than 80% drug release within the 60 min time frame. This indicated their
general suitability for formulating immediate- release solid oral formulations. The graph demonstrated in Figure 3 demonstrated a
correlation between particle size distribution of different grades of HiCelTM microcrystalline cellulose and the tablet properties. F1
containing HiCelTM 25M with a D-90 of 55.6 μm, exhibits the fastest disintegration time of 10.33 sec. This rapid disintegration
suggests a large surface area for water penetration and high porosity, lead to quick tablet breakdown. F2 containing HiCelTM 50M
with a D-90 of 132 μm and average weight of 265 mg showed increased dissolution time of 358 sec. Also, the F3 containing HiCelTM
90M with a D-90 of 210 μm resulted into slower disintegration time of 418 sec. F4 containing HiCelTM LP200 with a D-90 of 444
μm showed a disintegration time of 372 sec. As the particle size of microcrystalline cellulose is increased it had resulted into longer
disintegration time.
4. Conclusion
Evaluation of diclofenac sodium powder blends containing different HiCelTM Microcrystalline cellulose grades revealed significant
correlations between particle size distribution and micromeritic properties, compressibility and dissolution profiles. Fine grade,
HiCelTM 25M (F1) demonstrated poor flowability but excellent compressibility and rapid drug dissolution likely because of its fine
particle size and high surface area. In contrast, coarser grade HiCelTM LP200 (F4) exhibited superior flowability and fair
compressibility, leading to robust formulation with optimal dissolution characteristics. HiCelTM 50M (F2) also showed improved
flow and favorable dissolution, similar to F4. While HiCelTM 90M (F3) demonstrated fair tablet strength and disintegration time.
Overall, the study highlighted the critical role of microcrystalline cellulose particle size distribution in dictating the processability,
quality and performance of diclofenac sodium tablets, with HiCelTM LP200 emerging as a promising excipient for achieving a
balance of desirable micromeritics, manufacturing ease and formulation performance.
5. Acknowledgements
No funding was received for this research.
6. Conflict of interest
The authors state and confirm no conflict of interest.
7. References
1) Kumar V., Bhardwaj A., Singh N., Goyal K., Jindal S. A Review on Tablet Dosage Form: Recent Advancements with Special
Emphasis on Rapid Disintegrating Tablet. Asian J. Res. Pharm. Sci.,11(3):237-6. 2021.
2) Lachman L., Lieberman H.A., Kanig J.L., editors. The theory and practice of industrial pharmacy. Sec-III Pharmaceutical
Dosage Form, Chapter- Tablets (3rd ed.). Lea & Febiger; 1986.
3) Nofrerias I., Nardi-Ricart A., Suñé-Pou M., Suñé-Negre J.M., García-Montoya E., Pérez-Lozano P., Ticó J.R., Miñarro M.
Comparison between Microcrystalline Celluloses of different grades made by four manufacturers using the SeDeM diagram expert
system as a pharmaceutical characterization tool. Powder Technology. 342:780-788. 2019
4) Ait O.A., Harshe Y.M., Meunier V., Finke J.H., Influence of Process Parameters and Particle Size Distribution on Mechanical
Properties od Tablets. Chemie Ingenieur Technik. Vol-95:198-177. 2023.
5) Wünsch I., Finke J.H., John E., Kwade A. The influence of particle size on the application of compression and compaction
models for tableting. Int. J. Pharm., Vol-599. 2021.
6) Thoorens G., Krier F., Leclercq B., Carlin B., Evrard B. Microcrystalline cellulose, a direct compression binder in a quality by
design environment—A review. Int. J. Pharm., 473(1-2): 64-72. 2014
7) Roberts R.J., Rowe R.C. The effect of punch velocity on compaction of a variety of materials. J. Pharm. Pharmacol., 37(5):377-
380. 1985
8) Wang J., Wen H., Desai D. Lubrication in tablet formulations. Eur.J. Pharm., Vol-75(1);1-15. 2010.
9) Bolhuis G.K., Chowhan Z.T. Chapter Materials for direct compaction. Pharmaceutical Powder Compaction Technology. 419-
478. 1996.
10)United States Pharmacopoeia: General Chapter on bulk density <616>, 40-NF 35, 2018.
11)United States Pharmacopoeia: General Chapter on powder flow <1174>,40-NF 35, 2018.
12)United States Pharmacopoeia: General Chapter on powder flow <786>, 40-NF 35, 2018.
13)United States Pharmacopoeia: General Chapter on Uniformity of dosage units <905> 2024.
14)United States Pharmacopoeia: General Chapter on Tablet Breaking Force Determinations <1217> 2005.
15)United States Pharmacopoeia: General Chapter on Tablet Friability <1216> 2009.
16)United States Pharmacopoeia: General Chapter on Disintegration Time <701> 2008.
17)United States Pharmacopoeia: General Chapter on Dissolution Test <711> 2011.
18)Tomar M., Singh A.K., Sinha A.R. Physicochemical Parameter of Microcrystalline Cellulose and the most Acceptability in
Pharmaceutical Industries. J. Pharm. Biol. Sci. Vol-2 (4), 570-578. 2015.
19)Markl D., Zeitler J.A. A review of Disintegration Mechanisms and Measurement Techniques. J. Pharm. Res. 1;34(5):890–917.
2017.
20)Chaerunisaa A.Y., Sriwidodo S., Abdassah M. Microcrystalline Cellulose as Pharmaceutical Excipient. Pharm. Formul. Des. –
Recent Pract. 2019.
21)United States Pharmacopeia Guidelines for Dissolution Test. 44th edition, <711> 2011.
22)Sun J., Wang F., Sui Y., Zhennan S., Whai W., Wang C., Deng Y. Effect of particle size on solubility dissolution rate, and oral
bioavailability. National Library of Medicine. Int. J. Nanomed., 7:1521-30. 2012.
23)Kulthe V.V., Chaudhari P.D. Characterization of Etoricoxib Solid Dispersions Prepared by Spray Drying Technique. Res. J.
Pharm. Technol. Vol – 3 Page 1158-1166. 2010.